References
Liu, W., Wang, Z., You, R. et al. PLMSearch: Protein language model powers accurate and fast sequence search for remote homology. Nat Commun 15, 2775 (2024). https://doi.org/10.1038/s41467-024-46808-5Liu, W. et al. (2025). PLMSearch and PLMAlign: Protein Language Model (PLM)-Based Homologous Protein Sequence Search and Alignment. In: KC, D.B. (eds) Large Language Models (LLMs) in Protein Bioinformatics. Methods in Molecular Biology, vol 2941. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-4623-6_14
References
Liu, W., Wang, Z., You, R. et al. PLMSearch: Protein language model powers accurate and fast sequence search for remote homology. Nat Commun 15, 2775 (2024). https://doi.org/10.1038/s41467-024-46808-5Liu, W. et al. (2025). PLMSearch and PLMAlign: Protein Language Model (PLM)-Based Homologous Protein Sequence Search and Alignment. In: KC, D.B. (eds) Large Language Models (LLMs) in Protein Bioinformatics. Methods in Molecular Biology, vol 2941. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-4623-6_14
About the server
 |
- Source code: github.com/maovshao/PLMSearch
2. PLMAlign takes amino acid-level embeddings as input to obtain specific alignments and more refined similarity.
- Source code: github.com/maovshao/PLMAlign
- Webserver: dmiip.sjtu.edu.cn/PLMAlign
Note: If you have needs beyond this server, send your whole input file to us at maovshao@gmail.com for help.
Time cost
* With Swiss-Port (568K proteins) as target dataset.| Query Num | 1 | 10 | 100 |
| SS-predictor | 0.2 min | 0.5 min | 10.3 min |
| PLMSearch | 0.2 min | 1.1 min | 15.6 min |
| Query Num | 1 | 10 | 100 |
| SS-predictor | 1.6 min | 6.3 min | 60.2 min |
| PLMSearch | 2.3 min | 12.1 min | 114.6 min |
Tutorial
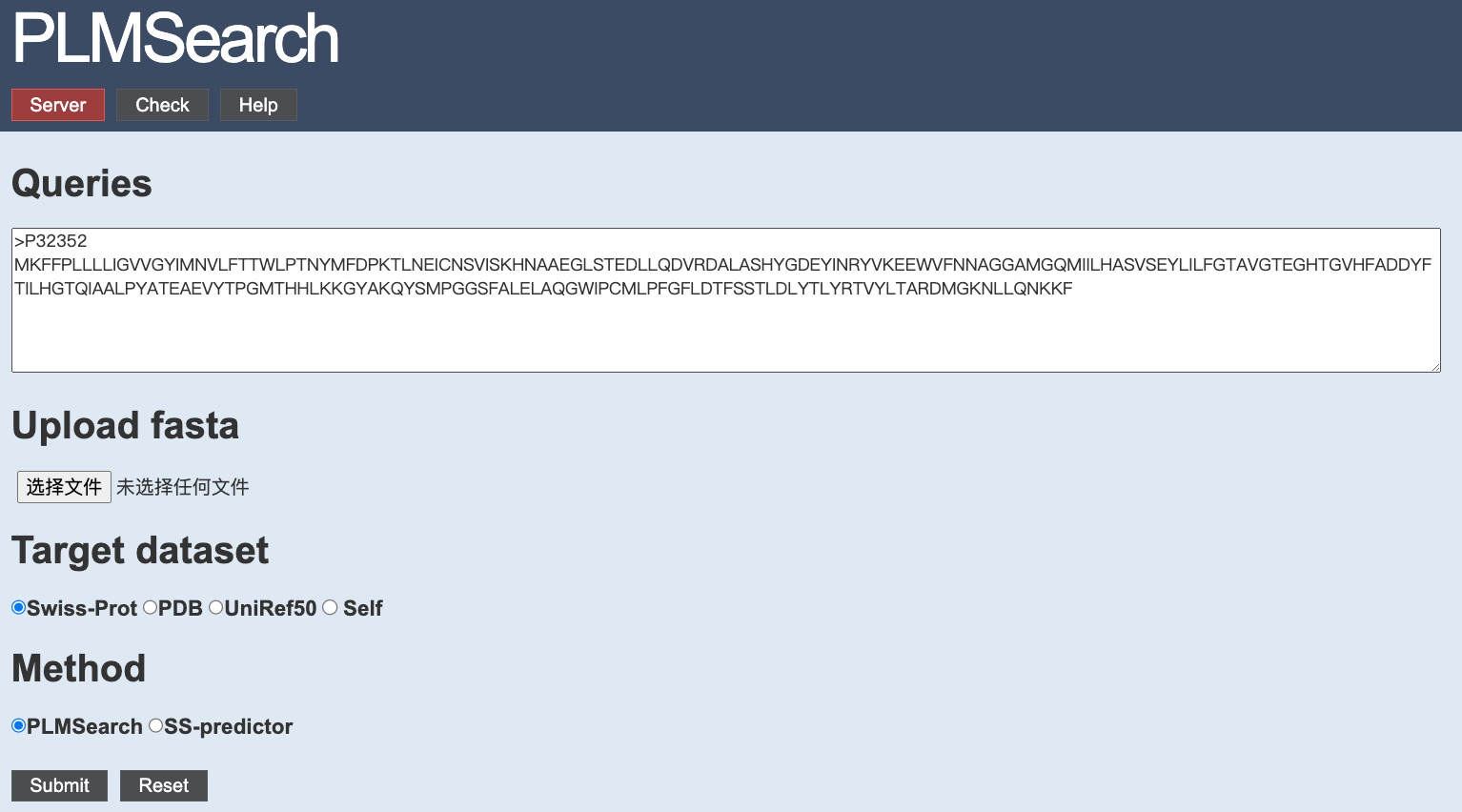 |
1. Input a protein sequence(s)
The sequences can either be typed directly into the text area, or uploaded from a file. If both, the server will only consider the sequence(s) in the uploaded file.Only the FASTA format is acceptable. All sequences have to be amino acids specified in a single letter code (ACDEFGHIKLMNPQRSTVWYVBZX*).
2. Choose a target dataset
You can choose Swiss-Port (568K proteins), PDB (680K proteins), UniRef50 (53.6M proteins), or the query dataset itself (Self).3. Choose a method
1. PLMSearch: Unlike SS-predictor below, PLMSearch is based on the pairs pre-filtered by PfamClan instead of searching all protein pairs from scratch.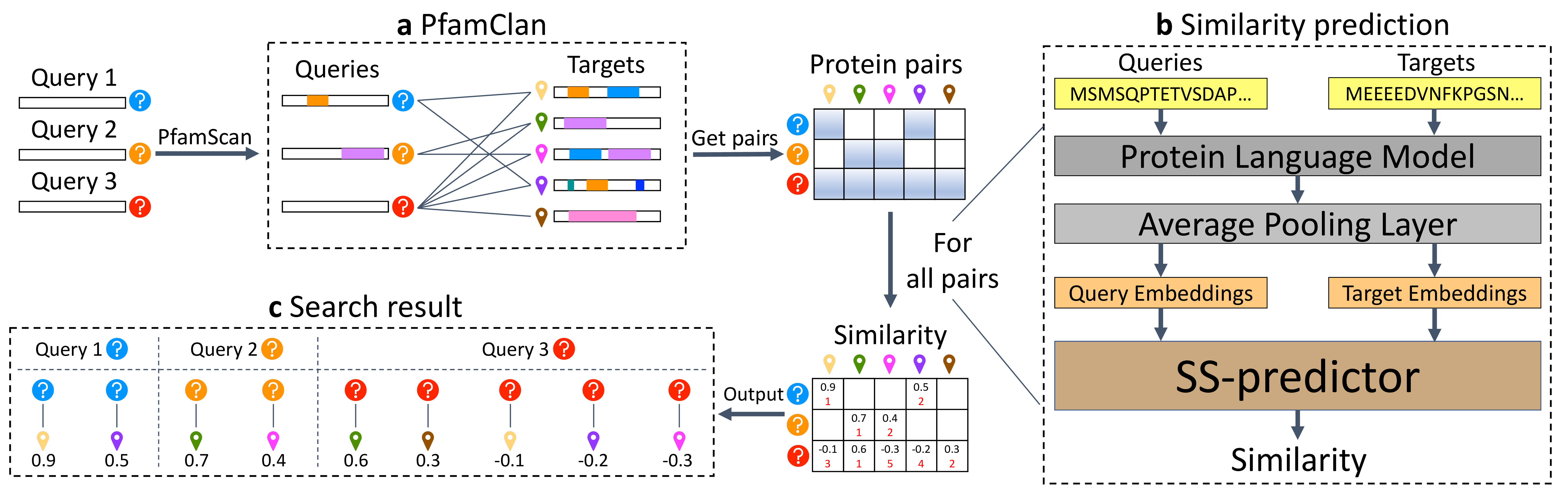 |
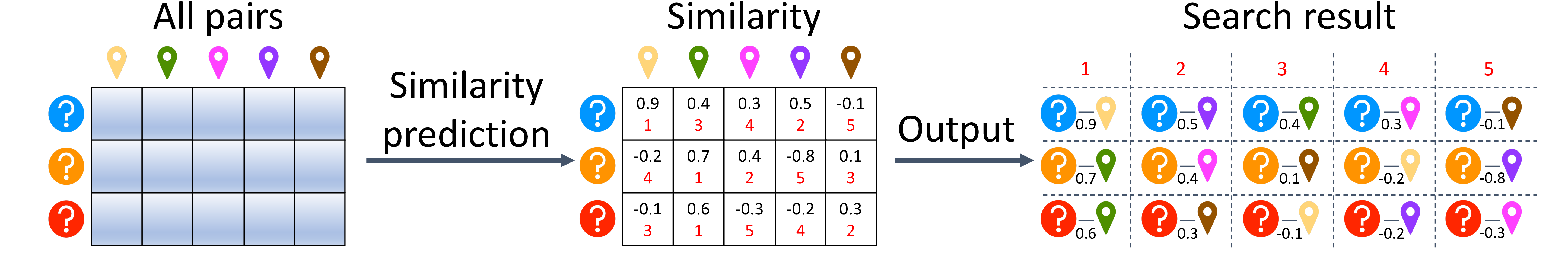 |
4. Submit
Press the "Submit" button to process the prediction. We will provide you a Job ID and a web link to the results.Use the "Reset" botton to clear all the inputs.
Obtain your search results at any time. Just "Check" your Job ID.
Interpreting the search result
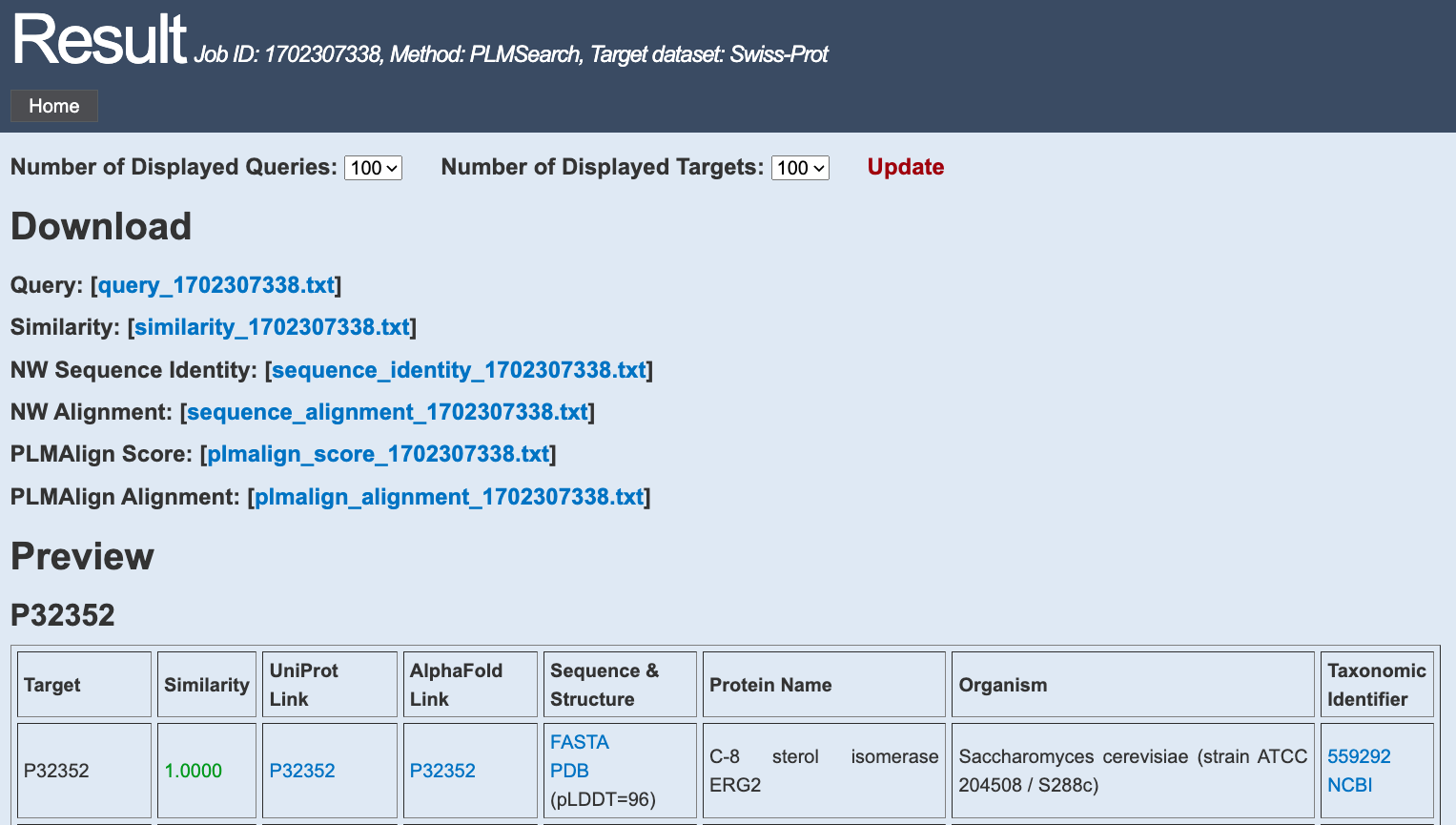 |
1. Download
Query: All your query proteins in the Fasta format.Similarity: The query-target pairs and their predicted similarity.
NW Sequence Identity: Sequence identity from Needleman-Wunsch algorithm.
NW Alignment: Sequence alignment from Needleman-Wunsch algorithm.
PLMAlign Score: Score from PLMAlign.
PLMAlign Alignment: Sequence alignment from PLMAlign.
Limitation
Query: The maximum supported number of query proteins for each submission is 100. If the number of proteins in your job exceeds, just divide them to separated jobs and submit them.Similarity: Only protein pairs with a similarity higher than 0.3 will be retained (0.5 for UniRef50). The maximum value of the total number of query-target pairs is 10,000.
In other words, the server will return the Top-K highest similarity for each protein in n queries, and K = 10,000 / n (If one query's actual search results are less than K, it will not be affected).
NW & PLMAlign: For each query, always return Top-10 alignment results.
Use our source code to implement more searches (github.com/maovshao/PLMSearch) and alignments (github.com/maovshao/PLMAlign).
2. Preview
Each table is presented for a query protein.Each row contains a target protein, arranged in descending order of similarity.
Some relevant information is provided for each target protein. Column by column from left to right: Target(Protein ID), Similarity, UniProt Link, AlphaFold Link, Sequence & Structure & (Avg. pLDDT), Protein Name, Organism, Taxonomic Identifier.
Note that depending on the target data set you choose, the style of the output table will be different.
Reference similarity
Researchers often wonder what the approximate similarity is for homologous protein pairs. Here, we answer this question by calculating the posterior probability for proteins at certain similarity having the same or different folds. The same or different folds are defined by SCOP.The posterior probability of proteins with a given similarity being in the same fold or different fold are shown below.
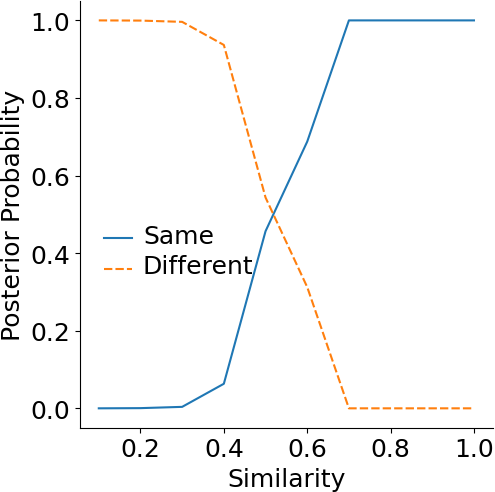 |
| Similarity | 0.1 | 0.3 | 0.5 | 0.7 | 0.9 |
| Posterior probability (same fold) | 0% | 0.37% | 45.65% | 100% | 100% |
| Posterior probability (different fold) | 100% | 99.62% | 54.34% | 0% | 0% |
Contact us
Please contact Shanfeng Zhu (zhusf@fudan.edu.cn) or Wei Liu (maovshao@gmail.com), if you have any question or comment about the server.User's feedback is quite important to us.
References
Liu, W., Wang, Z., You, R. et al. PLMSearch: Protein language model powers accurate and fast sequence search for remote homology. Nat Commun 15, 2775 (2024). https://doi.org/10.1038/s41467-024-46808-5Liu, W. et al. (2025). PLMSearch and PLMAlign: Protein Language Model (PLM)-Based Homologous Protein Sequence Search and Alignment. In: KC, D.B. (eds) Large Language Models (LLMs) in Protein Bioinformatics. Methods in Molecular Biology, vol 2941. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-4623-6_14